How To Design Safe Medical Products
This file type includes high-resolution graphics and schematics when applicable.
Designers new to the medical field sometimes are surprised to discover the lengths to which they must go before their medical devices can be considered safe for use. Young engineers, for example, frequently don’t expect that it takes five to ten times more effort to develop a device that is safe and complies with regulations than to develop a laboratory prototype. A device can only be considered safe after undergoing tests that prove its safety. So the safety discussion starts by devising the right tests that provide that proof.
Safety engineering principles emphasize that there are three aspects of making designs safe that are particularly important. They apply to the hardware, the software, and the user interface. Medical hardware uses a functional safety approach where two independent failures are not allowed to harm the patient. There are rules for designing software so the chances of harm arising from bugs are acceptably low. Finally, user interface design should employ usability rules that make the man-machine interface as safe as possible.
Engineers also are surprised to find that designing equipment to medical industry standards isn’t enough to guarantee that it is safe. It is understandable why this is so when you examine how standards come about. Standards are set by committees of experts. The standard-setting process is a political event; some committee members want strong requirements, some want weaker ones. It generally takes a long time to agree on specifics, so many standards are outdated by the time they publish. All in all, standards can’t hope to cover all risks. So designers must make up for the areas standards don’t cover by conducting a comprehensive risk analysis.
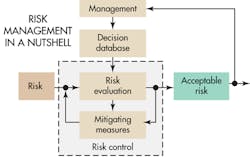
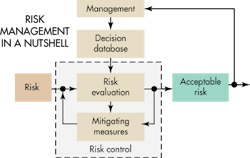
Risk management is actually a combination of several risk analysis methods that should let designers identify all relevant risks. In the medical field, ISO 14971:2007 specifies a process by which manufacturers can identify the hazards associated with medical devices, including in-vitro-diagnostic (IVD) medical devices, to estimate and evaluate the associated risks, to control these risks, and to monitor the effectiveness of the controls. The requirements of ISO 14971:2007 are applicable to all stages of the life cycle of a medical device.
However, medical devices have safety requirements that are less stringent than those for certain other product categories. For example, there are more requirements for making an airplane safe than for a medical device. The reason is that a medical device usually can only kill one person at a time while a commercial aircraft that isn’t safe might kill hundreds. Similarly, though medical devices must have designs that prevent two independent hardware failures from harming a patient, elevator designs must be safe in the event of three independent failures. All in all, the possibility of multiple lost lives brings with it stiffer requirements for safety.
Risk and safety
The task of developing medical devices that are safe boils down to identifying risks and then establishing the measures that give the confidence to say the risks are acceptable. Developers must judge the severity of potential harm and the probability that the harm occurs. Once developers have identified the unacceptable risks, their next step is to define safety measures to mitigate them.
For example, consider the case of an infusion pump. Its main function is to pump fluid. Potential hazards related to the pumping function include a wrong flow rate, infusing the wrong volume, an unintended start or stop of infusion, a buildup of excessive pressure, an infusion of air, and a reverse in the direction of flow. Designers would consider all these factors during development so the device could cause no harm in the case of a breakdown.
Fortunately there are standards that provide guidance on safety. The IEC 60601-1-2-24 pertains specifically to infusion pumps. Other standards pertain to other kinds of widely used medical equipment. An example is IEC 60601-2-16, which pertains to dialysis equipment. But, as stated before, standards aren’t enough to make devices safe. Designers must also conduct a formal risk analysis to determine requirements for the design of the device.
In that regard, ISO 14971 is a standard that details how manufacturers should conduct risk management to determine the safety of a medical device during the product life cycle. Such activity is required by higher level regulations and other quality standards such as ISO 13485.
The main standard for medical-device safety is IEC 60601-1 – Medical electrical equipment – Part 1: General requirements for basic safety and essential performance. Some countries also deviate from the standard under certain circumstances and sometimes use different versions of it. For example, the European and Canadian versions of the standard are identical to the IEC standard, but the U.S. version of the standard (ANSI/AAMI HA60601-1-11) excludes nursing homes from coverage. It also emphasizes usability requirements. Devices typically mandated to use the new standard include oxygen concentrators, body-worn nerve and muscle stimulators, beds, sleep-apnea monitors, and associated battery chargers prescribed for use at home.
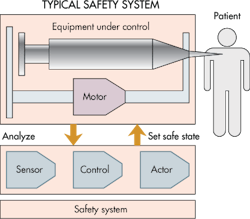
One of the principles of IEC 60601-1 is that a medical device must be safe in the case of a single fault. It defines a single fault as a failure of a safety system. Thus, one facet of designing a safe device is to imagine how a first failure in a safety system could endanger the patient, and then implement a safety system that still makes the device safe even in the event of a first failure.
One complicating factor is that the safety system has its own reliability level. Developers must establish what this level is. One approach to make safety systems reliable is to either use two redundant safety systems or use one system that is tested periodically to see if it is still functioning.
The basic approach is to go through every component in the device and figure out what happens if it fails. Each possible failure is acceptable if it is obvious and an operator can stop operations before the device can harm someone. For example, assume a safety system fails but the device continues to function properly. Designers must anticipate what happens in the event of a second safety-system failure after a certain time. When the safety system fails silently it no longer protects; still, the patient must be safe.
In the same vein, failures can be either systematic or random. Systematic failures are basically built-in design flaws. Examples include errors in the PCB layout, components used outside their specification, or unanticipated environmental conditions.
All software bugs are systematic failures; there are no random software failures. The effect of software failures might be random. For example, when a programmer doesn’t initialize a variable, its content on first use is random. This can cause a random effect at power on. The fact that the variable is not initialized is a systematic failure. Other examples of systematic failures in software include errors in the software specification, and errors in the operating system or compiler. Systematic errors in both hardware and software can be prevented through use of a robust development process.
Random errors, on the other hand, happen even though the design is correct and production is flawless. Random hardware errors can’t be predicted individually, they can only be described statistically. The general approach to controlling random errors is with redundant features or by adding self-testing or by adding a safety system that reacts in the event of a random failure. (Readers should note that a failure of a software storage medium is, in fact, a hardware and not a software error.)
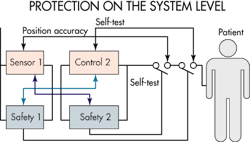
Designers typically use both redundancy and diversity as safety features. Redundancy is simply duplicating the same feature while diversity is the use of two different methods to deliver the same function. (The classic example is that of a seat belt and airbag protecting a car occupant from hitting the dashboard.) Diversity protects against random hardware errors as well as against some systematic failures. Redundancy, on the other hand, protects only against random hardware failures.
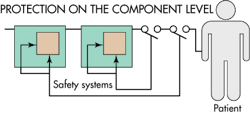
One of the questions that designers must decide is how much protection they must build in against random hardware failures. The answer depends on such factors as whether a first failure is a hazard and whether designers should assume there is a possibility of a second, third, or even more numerous failures. The main logic here is that hazards potentially able to kill multiple people at one strike demand more attention. The potential for harm will give guidance on how many independent failures designers must consider in the lifetime of the device. The potential for harm also determines the amount of redundant/diverse safety systems necessary. Normally medical devices kill one person, so designers must consider a maximum of two independent failures.
In general, designers must consider the possibility of ever-more unlikely events, the higher the risk of harm. For electrical medical devices, the IEC 60601-1 standard specifies that a combination of two independent failures should not be life threatening. This mandate expresses the concept of the single fault condition for medical devices. The principle is that a first failure should not cause a hazard. If the first failure is obvious to the operator, the operator stops using it and has it repaired. If the first failure can’t be detected, the designers must assume that a second failure will arise sometime later. They must also arrange the design so a combination of the first and second failures won’t cause a hazard.
Unfortunately the term “single-fault condition” can be misleading in the context of medical safety standards. It can suggest that designers need only assume that the device experiences only one failure. This is not correct.
Usually there is a time period after the first failure where the combination of a first and second failure is not allowed to be a hazard. For example, suppose the safety system has a first failure and undergoes a self-check within 24 hr that reveals the safety system is dead. That level of safety is acceptable in many medical systems. The assumption is that two independent safety-system failures would not arise within 24 hr of each other. Conversely, there is an unacceptable level of hazard if there is no self-check in the 24 hr after the first failure. In this case, the device either needs a self-check routine or a second safety system.
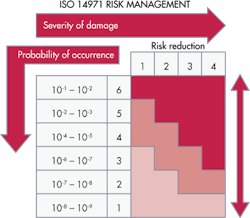
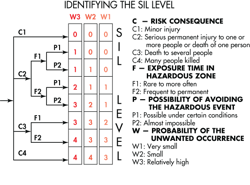
There is a progressive procedure for analyzing each hardware failure that could be dangerous. It is a risk graph spelled out in the IEC 61508 standard. It categorizes each fault condition in terms of a specific safety integrity level or SIL. Designers usually start by dividing risk consequences into four categories ranging from minor injury, serious injury, several deaths, and many deaths. They further subdivide risks according to the amount of exposure time to the hazard and the possibility of avoiding the hazardous event. Finally, they categorize the probability of the unwanted occurrence as very small, small, or relatively high.
Designers often start the safety analysis with a functional diagram of the product. This is an appropriate starting point because it’s possible to build in a high degree of safety on the level of system architecture. Designers typically treat each function as a black box. They then try to determine whether or not a specific black box is safe, or if it can be made safe by the introduction of safety systems. When they can’t determine the safety of a black box, they then open the box and go deeper, perhaps down to the level of individual components.
Fortunately designers in the U.S. need not rely on their own analysis of product functions to note safety red flags. The U.S. FDA maintains a market surveillance system that can give designers a heads-up on potential problem areas in medical devices. Any time a medical device has a failure, the manufacturer must report the details to the FDA. Alarms are raised if a specific device has a failure rate exceeding a certain threshold. Thus, medical-device engineers can consult this database to see what kinds of failures similar devices are experiencing.
Other countries have similar databases of medical device failures. However, their data tends to be less useful than that in the U.S. simply because individual countries each collect their own information. There is no central repository as yet for tabulating worldwide results.
The safety of software
Software for medical devices has its own standard, IEC 62304. It specifies life-cycle requirements for the development of medical software and software within medical devices. The standard spells out a risk-based decision model and defines testing requirements.
The primary principle for verifying software is to describe the function it is supposed to perform, then devise a test that verifies the software works as planned. The key lies in devising a test that is specific enough to identify all functions.
Unfortunately, the U.S. medical-device industry is not as advanced as it should be when it comes to implementing such procedures. In many cases, software descriptions tend to be ambiguous, and this condition causes several harmful side effects. For example, software engineers may develop something that has unintended functions. Equally bad, poor descriptions often prevent designers from devising all the tests that will expose harmful software bugs.
Additionally, many software engineers seem to have a too-high opinion of their own work. They seem to forget that numerous studies have shown software developers typically create between five and 10 bugs daily, a statistic that illustrates why an accurate description of software functions is essential.
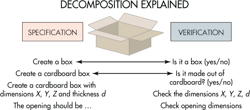
Software-safety analysis frequently employs a so-called V-model that is analogous to that widely used for visualizing the progression of system-development tasks. The model is named for its graphic depiction of how designers should decompose software requirements into ever-more-detailed specifications, then test and validate from the detailed levels up through to the system level.
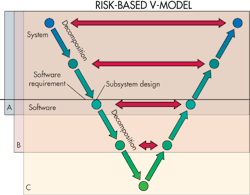
Most V-models divide decomposition steps into three levels. Top level is termed the system level. The lowest level represents the smallest software unit that can be 100% tested. The V-model represents the principle that designers must clearly describe software tasks in several levels of detail. There must be tests at every level that check that the software delivers its intended functions and identifies any bugs.
The software risk model defines three different levels of safety. Level A software is nonharmful if it fails. Level C software that fails can injure or kill someone. If software is neither A nor C, then it is level B by default. Categorizing software into one of the three levels helps determine how much testing is appropriate. Level A software just needs a system test. Level B tests must be detailed enough to check individual software modules. And as you might expect, most safety-system software in medical devices is at level C, which tests subsets of software code at the unit level.
The human element
It is no secret that appliances and instrumentation of all kinds are getting more complicated to operate. And medical instrumentation that is complicated is also prone to operator errors with potentially tragic consequences. Complicated instrumentation puts a demand on the operator’s intellectual ability. But users aren’t getting cleverer.
Unfortunately, standards for human-factor usability aren’t well developed. For example, one such document is IEC 62366 Annex D 1.4. It is weak in that it only supplies general guidelines about the steps necessary to identify risks of usage problems. It basically requires designers to analyze how users will interact with the device and implement risk-mitigating measures to avoid erroneous usage.
It can be quite expensive for manufacturers to analyze the ergonomics and usage of their equipment. One widely used technique is to round up 10 users, then ask them to say out loud what they are thinking as they operate the device. The whole set of interactions get recorded on video.
A typical finding in sessions like this is that only about 10% of instrument functions get used daily. The other 90% get used rarely or not at all. But it is not unusual to find often-used features buried in complicated menu structures with a huge potential for accidentally making an error.
However, there is no standard that requires an ergonomic analysis or user studies. These practices are simply recommended practices among firms that have experience developing medical equipment.
Voice Your Opinion!
To join the conversation, and become an exclusive member of Machine Design, create an account today!

Leaders relevant to this article: